
Density-functional tight-binding for phosphine-stabilized nanoscale gold clusters†
 ‡a
Jenica Marie L.
Madridejos,
‡b
Bálint
Aradi,
c
Bobby G.
Sumpter,
de
Gregory F.
Metha
*b
and
Stephan
Irle
*ae
‡a
Jenica Marie L.
Madridejos,
‡b
Bálint
Aradi,
c
Bobby G.
Sumpter,
de
Gregory F.
Metha
*b
and
Stephan
Irle
*ae
Abstract
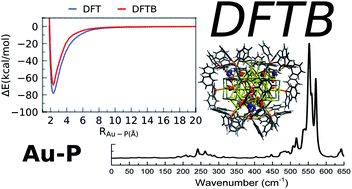
We report a parameterization of the second-order density-functional tight-binding (DFTB2) method for the quantum chemical simulation of phosphine-ligated nanoscale gold clusters, metalloids, and gold surfaces. Our parameterization extends the previously released DFTB2 “auorg” parameter set by connecting it to the electronic parameter of phosphorus in the “mio” parameter set. Although this connection could technically simply be accomplished by creating only the required additional Au–P repulsive potential, we found that the Au 6p and P 3d virtual atomic orbital energy levels exert a strong influence on the overall performance of the combined parameter set. Our optimized parameters are validated against density functional theory (DFT) geometries, ligand binding and cluster isomerization energies, ligand dissociation potential energy curves, and molecular orbital energies for relevant phosphine-ligated Aun clusters (n = 2–70), as well as selected experimental X-ray structures from the Cambridge Structural Database. In addition, we validate DFTB simulated far-IR spectra for several phosphine- and thiolate-ligated gold clusters against experimental and DFT spectra. The transferability of the parameter set is evaluated using DFT and DFTB potential energy surfaces resulting from the chemisorption of a PH3 molecule on the gold (111) surface. To demonstrate the potential of the DFTB method for quantum chemical simulations of metalloid gold clusters that are challenging for traditional DFT calculations, we report the predicted molecular geometry, electronic structure, ligand binding energy, and IR spectrum of Au108S24(PPh3)16.
 Please wait while we load your content...
Please wait while we load your content...

