
Overcoming the difficulties of predicting conformational polymorph energetics in molecular crystals via correlated wavefunction methods†

Abstract
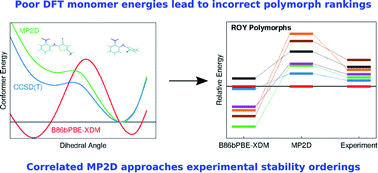
Molecular crystal structure prediction is increasingly being applied to study the solid form landscapes of larger, more flexible pharmaceutical molecules. Despite many successes in crystal structure prediction, van der Waals-inclusive density functional theory (DFT) methods exhibit serious failures predicting the polymorph stabilities for a number of systems exhibiting conformational polymorphism, where changes in intramolecular conformation lead to different intermolecular crystal packings. Here, the stabilities of the conformational polymorphs of o-acetamidobenzamide, ROY, and oxalyl dihydrazide are examined in detail. DFT functionals that have previously been very successful in crystal structure prediction perform poorly in all three systems, due primarily to the poor intramolecular conformational energies, but also due to the intermolecular description in oxalyl dihydrazide. In all three cases, a fragment-based dispersion-corrected second-order Møller–Plesset perturbation theory (MP2D) treatment of the crystals overcomes these difficulties and predicts conformational polymorph stabilities in good agreement with experiment. These results highlight the need for methods which go beyond current-generation DFT functionals to make crystal polymorph stability predictions truly reliable.
 Please wait while we load your content...
Please wait while we load your content...

