
Elucidating the role of solvents in acid catalyzed dehydration of biorenewable hydroxy-lactones†

Abstract
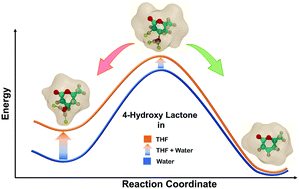
Owing to its low boiling point, tetrahydrofuran (THF) is an economical choice of solvent in biorenewables processing. Multifold acceleration in reaction rates is experimentally observed for Brønsted acid catalyzed reactions in THF, as compared to that in water. Herein, utilizing ab initio Car–Parrinello molecular dynamics (CPMD), classical molecular dynamics (MD) and density functional theory (DFT) simulations, a systematic theoretical framework is presented to explain the significant differences in the reactivity of Brønsted acid protons in THF as compared to that in water. The probe reaction of choice is the dehydration of 4-hydroxy-6-methyl lactone (HML) obtained from a biomass-derived substrate. Classical MD simulations elucidate the hydrogen bonding networks formed around the hydroxyl group of the reactant HML in explicit solvation environments of water and THF. The activation free energy barrier for the water removal step is calculated using CPMD–metadynamics simulations. In THF, the free energy barrier is 107 kJ mol−1, which is observed to be lower by 26 kJ mol−1 as compared to that in water. This significant difference in activation free energies for the dehydration step explains the difference in reactivity. Static DFT simulations further elaborated the effect of the first solvation shell around the hydroxyl substituent on describing the activation barriers of the dehydration reaction. While the solvent environment in DFT simulations is kept implicit in nature, few explicit molecules of THF and water are allowed to interact with the hydroxyl group and β-carbon of HML. The activation energy for the dehydration of HML is calculated to be 103 kJ mol−1 in the pure water environment. Akin to the difference in free energy barriers obtained from CPMD calculations, the activation energy calculated from DFT is observed to be 25 kJ mol−1 lower in THF as compared to that in water.
 Please wait while we load your content...
Please wait while we load your content...

