
Insight into atomically dispersed porous M–N–C single-site catalysts for electrochemical CO2 reduction†

Abstract
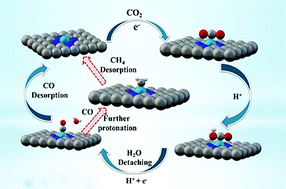
Transition metal single-site catalysts have unique activities for electrochemical CO2 reduction. However, the exact active center and reaction mechanism remain unclear due to a number of challenges in the controllable synthesis of single-atom catalysts (SACs) and defects in metal supports. Here we combine both experimental and theoretical calculations to systematically explore the mechanistic reaction path of selected transition metal single sites on nitrogen-doped porous carbon. Facile pyrolysis was employed to prepare a fullerene type carbon with 0.35 nm interlayer distances to support the family of M–N–C (M = Ni, Fe, Co and Cu). Experimentally, Ni and Fe outperform the other metals with high faradaic efficiency up to >97% and 86.8%, respectively. The theoretical calculations reveal that Ni–N–C exhibits optimum activity for CO2 reduction to CO at a higher overpotential because of the moderate *CO binding energy at the Ni site, which accommodates *COOH formation and *CO desorption. Furthermore, the strong binding energy of *CO on the Fe site enables the catalyst to reduce CO2 beyond CO. A remarkable current density of 17.6 mA cm−2 has been achieved with the Ni–N–C catalyst and a record of 5.74 s−1 TOF has been realized at −0.8 V vs. RHE for the Ni–N–C catalyst.
- This article is part of the themed collection: Top articles from NCNST
 Please wait while we load your content...
Please wait while we load your content...

