
Structural analysis of dehydrated gibbsite-based layered double hydroxides Li–Al–X (X = F−, Cl−, Br−, I−, OH−, NO3−, CO32−, and SO42−) by DFT calculations†

Abstract
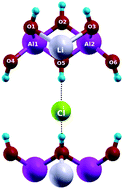
Ab initio calculations were performed in order to discuss the structural modifications derived from anion exchange in Li–Al–Cl LDH, a compound formed by the treatment of gibbsite with aqueous lithium chloride (LiCl). The Cl− ion in the LDH precursor was exchanged for a series of mono- and divalent anions, namely F−, Br−, OH−, I−, NO3−, CO32−, and SO42−. The XRD, NMR, and IR spectra were evaluated and direct comparisons with the experimental data showed good agreement. The interactions between the anions and the layers were evidenced by the charge density difference. It could be observed that the smaller the c parameter, the stronger the interaction between the layer and the anion. From pDOS analysis, it was possible to identify the basic sites in these LDHs. These sites may be responsible for the LDH selectivity in catalysis and for their adsorption properties.
- This article is part of the themed collection: Sustainability from intercalation compounds
 Please wait while we load your content...
Please wait while we load your content...

