
Development of transferable coarse-grained models of amino acids†

Abstract
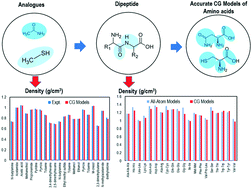
We have developed transferable coarse-grained (CG) models of the twenty standard amino acids, which can be used to perform molecular dynamics (MD) simulations of peptide amphiphiles (PAs) in the presence of explicit solvent. A 2 : 1 to 4 : 1 mapping scheme – in which a CG bead is comprised of two to four heavy atoms, respectively, and associated hydrogens – has been employed. Non-bonded parameters were optimized using the artificial neural network assisted particle swarm optimization (ANN-assisted PSO) method to reproduce experimental properties (density, surface tension, and heat of vaporization) of analogues of the side chains, termini, and backbone functional groups of the amino acids. The density (error <3.04%) and surface tension (error <7.38%) predicted by CG models were in good agreement with those of experimental properties. The peptide backbone is modeled with two charge neutral beads while amino acid side chains are modeled with one to three beads. Each terminus (N-terminus and C-terminus) is modeled as one charge neutral bead. Bonded parameters for the CG models were obtained from bond, angle, and dihedral distributions from AA MD simulations of dipeptides and/or tripeptides, which showed a reasonable agreement. Moreover, densities of these dipeptides and tripeptides calculated from AA MD simulations and CG models were in excellent agreement.
- This article is part of the themed collection: Bioinspired Materials
 Please wait while we load your content...
Please wait while we load your content...

