
Spin-state dependence of exchange–correlation holes
 a
and
Christoph R.
Jacob
*a
a
and
Christoph R.
Jacob
*a
Abstract
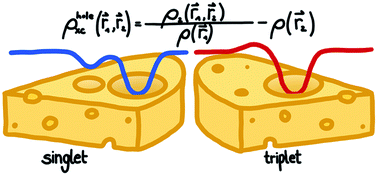
Applications of density-functional theory (DFT) in computational chemistry rely on an approximate exchange–correlation (xc) functional. However, existing approximations can fail dramatically for open-shell molecules, in particular for transition-metal complexes or radicals. Most importantly, predicting energy differences between different spin-states with approximate exchange–correlation functionals remains extremely challenging. Formally, it is known that the exact xc functional should be spin-state dependent, but none of the available approximations feature such an explicit spin-state dependence [C. R. Jacob and M. Reiher, Int. J. Quantum Chem., 2012, 112, 3661–3684]. Thus, to find novel approximations for the xc functional for open-shell systems, the development of spin-state dependent xc functionals appears to be a promising avenue. Here, we set out to shed light on the spin-state dependence of the xc functional by investigating the underlying xc holes, which we extract from configuration interaction calculations for model systems. We analyze the similarities and differences between the xc holes of the lowest-energy singlet and triplet states of the dihydrogen molecule, the helium atom, and the lithium dimer. To shed further light on the spin-state dependence of these xc holes we also discuss exact conditions that can be derived from the spin structure of the reduced two-electron density matrix. Altogether, our results suggest several possible routes towards the construction of explicitly spin-state dependent approximations for the xc functional.
- This article is part of the themed collection: New horizons in density functional theory
 Please wait while we load your content...
Please wait while we load your content...

