
Gauging aromatic conjugation and charge delocalization in the aryl silanes PhnSiH4−n (n = 0–4), with silicon K-edge XAS and TDDFT†

Abstract
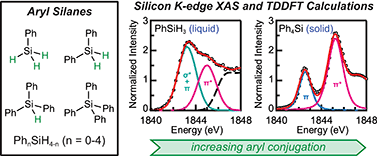
Si K-edge X-ray absorption spectra (XAS) have been measured experimentally and calculated using time-dependent density functional theory (TDDFT) to investigate electronic structure in aryl silanes, PhnSiH4−n (n = 0–4). Adding aryl groups to SiH4 splits the Si–H σ-antibonding orbitals into new orbitals with Si–Ph π-bonding (πb) and π-antibonding (π*) character. Greater aryl substitution is reflected by increasingly intense Si 1s → πb and Si 1s → π* transitions, and weaker transitions into the Si–H and Si–C σ* orbitals. These observations are consistent with known trends in the hydride donor ability of aryl silanes, which is driven in part by the composition of the LUMOs and the accessibility of pathways for electron delocalization through aromatic conjugation. Methodology developed for liquid-phase Si K-edge XAS measurements on PhSiH3 and Ph2SiH2 will enable dynamic studies of chemical transformations involving silicon-containing catalysts, intermediates, and substrates.
 Please wait while we load your content...
Please wait while we load your content...

