
Exploring direct and hydrogen-assisted CO activation on iridium surfaces – surface dependent activity†
 ab
Haijun
Jiao
*ad
ab
Haijun
Jiao
*ad
Abstract
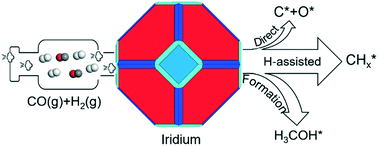
To understand the mechanisms of CO activation and conversion on noble metal surfaces, direct dissociation, H-assisted activation and successive hydrogenation of CO to methanol have been computed on flat Ir(111) and Ir(100), corrugated Ir(110) and Ir(210) and stepped Ir(311) and Ir(221) surfaces. On these surfaces, CO strongly prefers top adsorption sites and has much stronger adsorption than at high coordination sites. The CO direct dissociation barrier is higher than the CO desorption energy and the reaction is very endothermic, and thus not accessible, while H-assisted CO activation is more preferred kinetically. On the H-assisted pathway, the minimum energy path is CO* → HCO* → HCOH* → HC* + OH* on the Ir(111) and Ir(100) surfaces, CO* → HCO* → HCOH* → H2COH* → H2C* + OH* on the Ir(110), Ir(210) and Ir(311) surfaces and CO* → HCO* → H2CO* → H2COH* → H2C* + OH* on the Ir(221) surface, respectively. Interestingly, methanol formation has close effective barriers with the respective C–O dissociation of the H-assisted pathway, indicating that CO can not only be hydrogenated to methanol but also form 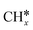 via H-assisted dissociation on the Ir surfaces. The adsorptions of surface intermediates on high index surfaces are not always stronger than those on low index surfaces, indicating that the high index surfaces should not be always blocked by surface intermediates. The behaviors of CO in adsorption, dissociation and activation on Ir surfaces are compared with those on low and high index surfaces of Fe, Co, Ni and Ru metals.
via H-assisted dissociation on the Ir surfaces. The adsorptions of surface intermediates on high index surfaces are not always stronger than those on low index surfaces, indicating that the high index surfaces should not be always blocked by surface intermediates. The behaviors of CO in adsorption, dissociation and activation on Ir surfaces are compared with those on low and high index surfaces of Fe, Co, Ni and Ru metals.
 Please wait while we load your content...
Please wait while we load your content...

