
Method for the accurate prediction of electron transfer potentials using an effective absolute potential†

Abstract
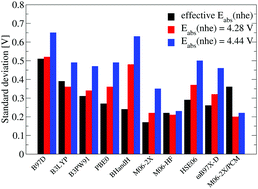
A protocol for the accurate computation of electron transfer (ET) potentials from ab initio and density functional theory (DFT) calculations is described. The method relies on experimental pKa values, which can be measured accurately, to compute a computational setup dependent effective absolute potential. The effective absolute potentials calculated using this protocol display strong variations between the different computational setups and deviate in several cases significantly from the “generally accepted” value of 4.28 V. The most accurate estimate, obtained from CCSD(T)/aug-ccpvqz, indicates an absolute potential of 4.14 V for the normal hydrogen electrode (nhe) in water. Using the effective absolute potential in combination with CCSD(T) and a moderately sized basis, we are able to predict ET potentials accurately for a test set of small organic molecules (σ = 0.13 V). Similarly we find the effective absolute potential method to perform equally good or better for all considered DFT functionals compared to using one of the literature values for the absolute potential. For, M06-2X, which comprises the most accurate DFT method, standard deviation of 0.18 V is obtained. This improved performance is a result of using the most appropriate effective absolute potential for a given method.
 Please wait while we load your content...
Please wait while we load your content...

