
Discovery of potent inhibitors for SARS-CoV-2's main protease by ligand-based/structure-based virtual screening, MD simulations, and binding energy calculations†

Abstract
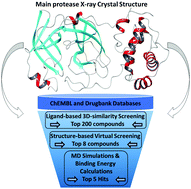
COVID-19 has caused lockdowns all over the world in early 2020, as a global pandemic. Both theoretical and experimental efforts are seeking to find an effective treatment to suppress the virus. In silico drug design can play a vital role in identifying promising drug candidates against COVID-19. Herein, we focused on the main protease of SARS-CoV-2 that has crucial biological functions in the virus. We performed a ligand-based virtual screening followed by a docking screening for testing approved drugs and bioactive compounds listed in the DrugBank and ChEMBL databases. The top 8 docking results were advanced to all-atom MD simulations to study the relative stability of the protein–ligand interactions. MD simulations support that the catalytic residue, His41, has a neutral side chain with a protonated delta position. An absolute binding energy (ΔG) of −42 kJ mol−1 for the protein–ligand (Mpro-N3) complex has been calculated using the potential-of-mean-force (geometrical) approach. Furthermore, the relative binding energies were computed for the top docking results. Our results suggest several promising approved and bioactive inhibitors of SARS-CoV-2 Mpro as follows: a bioactive compound, ChEMBL275592, which has the best MM/GBSA binding energy; the second-best compound, montelukast, is an approved drug used in the treatment of asthma and allergic rhinitis; the third-best compound, ChEMBL288347, is a bioactive compound. Bromocriptine and saquinavir are other approved drugs that also demonstrate stability in the active site of Mpro, albeit their relative binding energies are low compared to the N3 inhibitor. This study provides useful insights into de novo protein design and novel inhibitor development, which could reduce the cost and time required for the discovery of a potent drug to combat SARS-CoV-2.
 Please wait while we load your content...
Please wait while we load your content...

