
An approach to calculate the free energy changes of surface reactions using free energy decomposition on ab initio brute-force molecular dynamics trajectories†
 *a
*a
Abstract
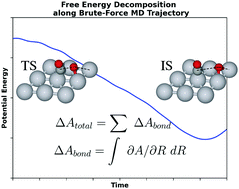
To understand the mechanisms and kinetics of catalytic reactions in heterogeneous catalysis, ab initio molecular dynamics is one of the powerful methods used to explore the free energy surface (FES) of surface elementary steps. The most significant aspect of performing such calculations is to choose the specific collective variable (CV) of the reaction. Here, we take CO oxidation on Pt(111) at 300 K as an example to demonstrate the protocol of selecting CVs guided by the free energy decomposition which quantifies individual bond free energy contributions. The basic concept is to conduct the brute-force molecular dynamics initiated from the transition state on the FES, which is refined from the one on the potential energy surface, to generate the reaction path at a finite temperature. The validity of this reaction path is further demonstrated by a 2-D free energy landscape spanned by the path-CV. By choosing CVs including other bond distances, we find that CO oxidation cannot be well understood by umbrella sampling or constrained molecular dynamics (CMD) solely along the OC–O bond distance. The free energy decomposition analysis suggests that not only the OC–O bond but also two O–Pt bonds are responsible for the free energy change. The further CMD simulations along selected CVs based on the insights from our protocol capture different reaction stages and give solid estimations of free energy barriers.
 Please wait while we load your content...
Please wait while we load your content...

