
Carbon phosphide nanosheets and nanoribbons: insights on modulating their electronic properties by first principles calculations†

Abstract
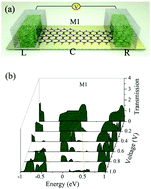
A carbon phosphide (CP) monolayer, a 2D structure derived from the same 3-fold coordination found both in graphene and phosphorene, has been successfully synthesized in an experiment recently. In this paper, we investigated the modulation of electronic structures and transport characteristics of 2D nanosheets and quasi-1D nanoribbons of CP nanomaterials in the α-phase by using first-principles density functional theory simulation. The calculated band structures show that the band gap of 2D CP nanosheets progressively increases as the uniform biaxial strain changes from compression to stretching. However, the biaxial strain cannot change the indirect band gap behavior of the original 2D CP nanosheet. In addition, the band structures of quasi-1D nanoribbons with different styles of H-passivated zigzag edges have also been studied. The results show that the H-passivated zigzag PC ribbons with two P edges are semiconductors with indirect band gaps, and the gaps decrease with increasing width of ribbons. However, the H-passivated CP nanoribbons with one P-atom terminated edge in combination with one P-atom edge, and H-passivated CC nanoribbons with two C-atom terminated edges display metallic behaviors. The semi-conductive or metallic behaviors of zigzag CP nanoribbons can be explained by presenting the wave function of their energy band around the Fermi level. Finally, the electronic transport properties of different CP nanoribbon based nanojunctions are studied in which arise the interesting negative differential resistance or rectification effects in their current–voltage characteristic curves.
 Please wait while we load your content...
Please wait while we load your content...

