
First-principles calculations of anharmonic and deuteration effects on the photophysical properties of polyacenes and porphyrinoids

Abstract
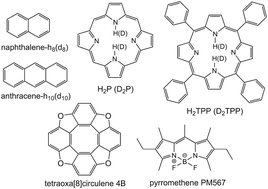
A new method for calculating internal conversion rate constants (![[k with combining low line]](https://www.rsc.org/images/entities/i_char_006b_0332.gif) IC), including anharmonic effects and using the Lagrangian multiplier technique, is proposed. The deuteration effect on IC is investigated for naphthalene, anthracene, free-base porphyrin (H2P) and tetraphenylporphyrin (H2TPP). The results show that anharmonic effects are important when calculating IC for transitions between electronic states that are energetically separated (ΔE) by more than 20 000–25 000 cm−1. Anharmonic effects are also important when ΔE < 20 000–25 000 cm−1 and when the accepting modes are X–H stretching vibrations with a frequency larger than 2000 cm−1. The calculations show that there is mixing between the S1 and S2 states of naphthalene induced by non-adiabatic interactions. The non-adiabatic interaction matrix element between the S1 and S2 states is 250 cm−1 and 50 cm−1 for the normal and fully deuterated naphthalene structure and this difference significantly affects the estimated fluorescence quantum yield. Besides aromatic hydrocarbons H2P and H2TPP, the IC rate constant is also calculated for pyrometene (PM567) and tetraoxa[8]circulene (4B) with a detailed analysis of the effect of the vibrational anharmonicity.
IC), including anharmonic effects and using the Lagrangian multiplier technique, is proposed. The deuteration effect on IC is investigated for naphthalene, anthracene, free-base porphyrin (H2P) and tetraphenylporphyrin (H2TPP). The results show that anharmonic effects are important when calculating IC for transitions between electronic states that are energetically separated (ΔE) by more than 20 000–25 000 cm−1. Anharmonic effects are also important when ΔE < 20 000–25 000 cm−1 and when the accepting modes are X–H stretching vibrations with a frequency larger than 2000 cm−1. The calculations show that there is mixing between the S1 and S2 states of naphthalene induced by non-adiabatic interactions. The non-adiabatic interaction matrix element between the S1 and S2 states is 250 cm−1 and 50 cm−1 for the normal and fully deuterated naphthalene structure and this difference significantly affects the estimated fluorescence quantum yield. Besides aromatic hydrocarbons H2P and H2TPP, the IC rate constant is also calculated for pyrometene (PM567) and tetraoxa[8]circulene (4B) with a detailed analysis of the effect of the vibrational anharmonicity.
- This article is part of the themed collection: 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

