
What is the driving force behind molecular triangles and their guests? A quantum chemical perspective about host–guest interactions†
 *a
Maurício J.
Piotrowski,
b
Àlvaro
Muñoz-Castro,
c
Renato L. T.
Parreira
d
and
Giovanni F.
Caramori
e
*a
Maurício J.
Piotrowski,
b
Àlvaro
Muñoz-Castro,
c
Renato L. T.
Parreira
d
and
Giovanni F.
Caramori
e
Abstract
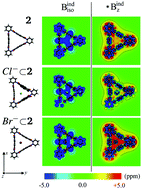
The physical nature of host–guest (HG) interactions occurring between molecular triangles and linear anions was explored using density functional theory (DFT) calculations combined with energy decomposition analyses (EDA), nuclear independent chemical shift (NICS), and non-covalent interaction index (NCI). We demonstrate that: (i) in addition to the host being significantly rigid, the strain energies are not negligible, especially for host 2; (ii) halogen anions interact mainly by electrostatic forces (ΔEelst > ΔEorbtot > ΔEdisp), meanwhile; (iii) trihalogen anions interact mostly by dispersion forces (ΔEdisp > ΔEelst ≈ ΔEorbtot). The NICS and NCI calculations corroborate the idea that HG interactions are considerably mediated through dispersion terms, and also indicate an antiaromatic character inside the host walls.
 Please wait while we load your content...
Please wait while we load your content...

