
Molecular-level insights into the structures, dynamics, and hydrogen bonds of ethylammonium nitrate protic ionic liquid at the liquid–vacuum interface†
 *a
*a
Abstract
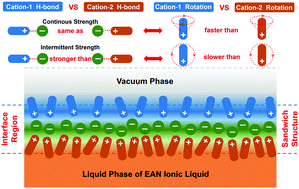
A series of molecular dynamics simulations have been used to systematically explore the structures, dynamics and hydrogen bonds (HBs) of ethylammonium nitrate (EAN) protic ionic liquid (IL) and their mutual relationship at the liquid–vacuum interface. The simulation results clearly demonstrate that there exists a sandwich structure at the interface, with the double-layer of the EA+ cations on both sides and one intercalated layer of the NO3− anions in the middle. Wherein, the outermost cation layer prefers the orientation with the CH3 groups pointing to the vacuum phase due to the hydrophobic interactions, while the CH3 groups in the second layer direct to the bulk liquid phase owing to the HB formation between their NH3+ groups and the intercalated NO3− anions in the middle layer. On the other hand, the continuous HB strength of the cations in the outermost layer (denoted as Cation-1) is found to be almost identical with the counterpart of the cations in the second layer (denoted as Cation-2), whereas the intermittent HB strength of Cation-1 is much larger than that of Cation-2 at all temperatures. Furthermore, the rotational motion of Cation-1 with the normal vector of the C–C–N plane in the cation is faster than that of Cation-2 with the same vector, resulting from more free space in the outermost layer. On the contrary, the rotational motion of Cation-1 with the vector from the mass center of the cation to its N atom is much slower than that of Cation-2 with the same vector, which can be attributed to the combined effects of the stronger intermittent HBs of Cation-1 and the hydrophobic interactions of its CH3 group in the outermost layer.
 Please wait while we load your content...
Please wait while we load your content...

