
A vibrational spectroscopic and computational study of gaseous protonated and alkali metal cationized G–C base pairs†
 b
and
Travis D.
Fridgen
*a
b
and
Travis D.
Fridgen
*a
Abstract
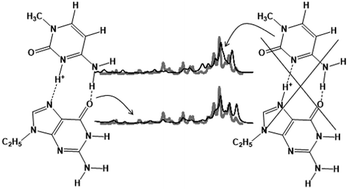
The structures and properties of metal cationized complexes of 9-ethylguanine (9eG) and 1-methylcytosine (1mC), (9eG:1mC)M+, where M+ = Li+, Na+, K+, Rb+, Cs+ as well as the protonated complex, (9eG:1mC)H+, have been studied using a combination of IRMPD spectroscopy and computational methods. For (9eG:1mC)H+, the dominant structure is a Hoogsteen type complex with the proton covalently bound to N3 of 1mC despite this being the third best protonation site of the two bases; based on proton affinities N7 of 9eG should be protonated. However, this structural oddity can be explained considering both the number of hydrogen bonds that can be formed when N3 of 1mC is protonated as well as the strong ion-induced dipole interaction that exists between an N3 protonated 1mC and 9eG due to the higher polarizability of 9eG. The anomalous dissociation of (9eG:1mC)H+, forming much more (1mC)H+ than would be predicted based on the computed thermochemistry, can be explained as being due to the structural oddity of the protonation site and that the barrier to proton transfer from N3 of 1mC to N7 of 9eG grows dramatically as the base pair begins to dissociate. For the (9eG:1mC)M+; M = Li+, Na+, K+, Rb+, Cs+ complexes, single unique structures could not be assigned. However, the experimental spectra were consistent with the computed spectra. For (9eG:1mC)Li+, the lowest energy structure is one in which Li+ is bound to O6 of 9eG and both O2 and N3 of 1mC; there is also an interbase hydrogen bond from the amine of 1mC to N7 of 9eG. For Na+, K+, and Rb+, similar binding of the metal cation to 1mC is calculated but, unlike Li+, the lowest energy structure is one in which the metal cation is bound to N7 of 9eG; there is also an interbase hydrogen bond between the amine of 1mC and the carbonyl of 9eG. The lowest energy structure for the Cs complex is the Watson–Crick type base pairing with Cs+ binding only to 9eG through O6 and N7 and with three hydrogen bonds between 9eG and 1mC. It also interesting to note that the Watson–Crick base pairing structure gets lower in Gibbs energy relative to the lowest energy complexes as the metal gets larger. This indicates that the smaller, more densely charged cations have a greater propensity to interfere with Watson–Crick base pairing than do the larger, less densely charged metal cations.
 Please wait while we load your content...
Please wait while we load your content...

