
Modelling the bulk properties of ambient pressure polymorphs of zirconia†
 *ab
Matthew G.
Quesne
ab
and
*ab
Matthew G.
Quesne
ab
and
Abstract
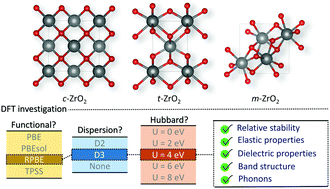
We report a detailed survey of the calculated bulk properties of zirconia using GGA and meta-GGA functionals (PBE, PBEsol, RPBE, and TPSS), dispersion (Grimme's D2 and D3 approach), and on-site Coulomb repulsion correction (U = 2–8 eV). Structural, elastic, mechanical, and dielectric properties, as well as energetics, electronic structure, and phonon dispersion curves were computed and compared to previous investigations to identify the best DFT approach for a consistent in silico description of zirconia polymorphs. In general, inclusion of dispersion corrections led to only small changes in the calculated properties, whereas DFT+U (U = 2 or 4 eV) reduced the deviations of calculated properties from the experimental results, although deterioration of the structure and relative stabilities may be observed in some cases. Standard PBEsol, RPBE+U, and PBE+U were the best methodologies for a simultaneous description of the three polymorphs of ZrO2. RPBE+U, however, was the only functional to conserve the distinct structures and stabilities of c-, t-, and m-ZrO2 when U = 4 eV was used, resulting in the best in silico replication of the band gaps of ZrO2, whilst outperforming the other methodologies in the description of elastic, mechanical, and dielectric properties of this material. Overall, these results provide insight into the most appropriate DFT methodology for in silico investigations of ZrO2, and show that simultaneous description of all three ambient pressure zirconia polymorphs by DFT techniques with acceptable levels of accuracy can be achieved only when the correct choice of methodology is applied.
- This article is part of the themed collection: Celebrating our 2020 Prize and Award winners
 Please wait while we load your content...
Please wait while we load your content...

