
DFT study of various tungstates for photocatalytic water splitting†

Abstract
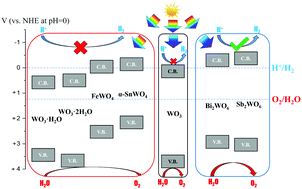
Tungsten oxide (WO3) is a promising photocatalytic material, but it has some limitations on its optoelectronic properties. Compared with binary materials, ternary compounds provide a much greater variety of compositions and hence properties, which can be tuned to suit particular applications. In this work, the effect of introducing a second metal cation into tungsten oxide is studied by Density Functional Theory (DFT) calculations. The compounds investigated include AWO4 tungstates (A = Sn, Fe), M2WO6 tungstates (M = Bi, Sb), tungstite (WO3·H2O) and hydrotungstite (WO3·2H2O). The tungstates studied are found to have either a small band gap (SnWO4, FeWO4, WO3·H2O and WO3·2H2O), and thus potentially improved visible-light activity compared with WO3, or a more negative conduction band edge than WO3 (Bi2WO6, Sb2WO6), which means they may be able to achieve overall water splitting, in contrast to WO3. The band gap narrowing and the band edge changes are attributed to the introduction of new electronic states due to the second metal cation, as well as structural changes, particularly a larger spacing between layers of WO6 octahedra. All the materials studied have a relative high static dielectric constant (εr > 10), allowing for exciton dissociation, and a small enough electron effective mass (me* < 0.5m0) along at least one direction for carrier diffusion. The performance of all the compounds is likely to be limited by poor hole mobility, except for Sb2WO6 and the hydrated compounds which also have a relatively small hole effective mass (mh* < 0.5m0). Through this comparative study, the key trends in properties as a function of composition for a family of complex materials have been identified, allowing appropriate compositions to be selected and tuned for specific applications.
 Please wait while we load your content...
Please wait while we load your content...

