
Time-dependent DFT study of the K-edge spectra of vanadium and titanium complexes: effects of chloride ligands on pre-edge features†

Abstract
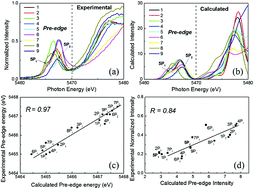
X-ray absorption near edge structures (XANES) of vanadium and titanium complexes were investigated with time-dependent density functional theory (TDDFT). In particular, observed characteristic K-edge features in the presence of chloride ligands were assigned. Although TDDFT includes a large systematic error attributed to the 1s core energy levels of transition metals, pre-edge spectral shapes could be reproduced by a simple energy shift in the calculated excitation energies. The doublet peak in the pre-edge region was assigned to dipole-allowed transitions from 1s to 3d + 4p hybridized orbitals, while a characteristic shoulder peak in the chloride complex was assigned to excitations of chloride 4p orbitals. A similar but weak absorption band was computed for the methyl complex as excitation to C–H σ* orbitals. However, because these excitations were highly dependent on the direction of the C–H bonds, the shoulder peak was not experimentally observed because of methyl free rotation. Hence, the intensity of the shoulder peak was proportional to the number of chloride ligands unless other ligands contribute to this energy region and, therefore, could be used to detect the presence or absence of chloride ligands in unknown compounds, such as reaction intermediates.
 Please wait while we load your content...
Please wait while we load your content...

