
An accurate potential energy surface and ring polymer molecular dynamics study of the Cl + CH4 → HCl + CH3 reaction†

Abstract
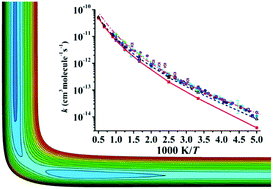
The reaction Cl + CH4 → HCl + CH3, a prototypical bimolecular reaction, has been established as an important proving ground for studying chemical reaction kinetics and dynamics of polyatomic molecules. In this work, a globally accurate full-dimensional potential energy surface (PES) for this reaction is developed by using the permutationally invariant polynomial neural network (PIP-NN) approach based on 74 000 points calculated at the level of the explicitly correlated unrestricted coupled cluster single, double, and perturbative triple level with the augmented correlation corrected valence triple-zeta basis set (UCCSD(T)-F12a/AVTZ). For points in the entrance channel, spin–orbit corrections stemming from Cl(2P) are determined at the level of complete active space self-consistent field (CASSCF) with the AVTZ basis set. With this PES, thermal rate coefficients and kinetic isotope effects are computed for reactions Cl + CH4 → HCl + CH3 and Cl + CD4 → DCl + CD3 using the ring polymer molecular dynamics (RPMD) method, which can provide reliable estimations for thermal rate coefficients effectively. Generally, the agreement with the scattered experimental results is reasonably satisfactory.
- This article is part of the themed collection: Emerging AI Approaches in Physical Chemistry
 Please wait while we load your content...
Please wait while we load your content...

