
Towards red-light o-carborane derivatives with both aggregation induced emission and thermally activated delayed fluorescence combining quantum chemistry calculation with molecular dynamics simulation†
 *a
Zhong-Min
Su
*acd
*a
Zhong-Min
Su
*acd
Abstract
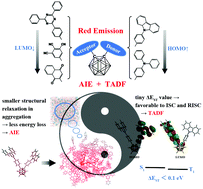
Two o-carborane derivatives 1 and 2 exhibiting both aggregation induced emission (AIE) and thermally activated delayed fluorescence (TADF) properties were investigated by combining density functional theory with molecular dynamics simulation. The reason for their TADF property was explored. Besides, taking 1 as an example, the AIE phenomenon was also probed by simulating the aggregation process and comparing the photophysical properties of the molecules in different aggregation states (including crystals, water, and films) with the isolated molecule. The results manifest that the separated orbital occupations of the HOMO and LUMO at the respective donor and acceptor units result in a small singlet–triplet energy gap and a favorable TADF feature, which is similar to many typical TADF molecules. In the AIE phenomenon, a much smaller structural change of donor and acceptor units between S1 and S0 states was experienced for the molecules in all aggregate states compared with that for the isolated molecule, decreasing the energy dissipation substantially and enhancing the luminescence efficiency. In addition, o-carborane is conjectured to help in the free rotation of donor and acceptor units for the isolated molecule, which may contribute to AIE. What's more, on the basis of 1 and 2, we designed 3–6 by means of replacing the triphenyltriazine group with other electron-withdrawing groups having lower LUMO energy levels, aiming to obtain this kind of red light emitting material. The results indeed proved our assumption that 3–6 not only present the feature of red light emission but also have both TADF and AIE properties in films.
 Please wait while we load your content...
Please wait while we load your content...

