
A generalized deep learning approach for local structure identification in molecular simulations†

Abstract
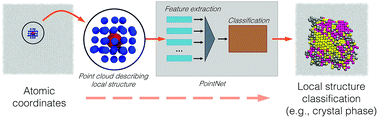
Identifying local structure in molecular simulations is of utmost importance. The most common existing approach to identify local structure is to calculate some geometrical quantity referred to as an order parameter. In simple cases order parameters are physically intuitive and trivial to develop (e.g., ion-pair distance), however in most cases, order parameter development becomes a much more difficult endeavor (e.g., crystal structure identification). Using ideas from computer vision, we adapt a specific type of neural network called a PointNet to identify local structural environments in molecular simulations. A primary challenge in applying machine learning techniques to simulation is selecting the appropriate input features. This challenge is system-specific and requires significant human input and intuition. In contrast, our approach is a generic framework that requires no system-specific feature engineering and operates on the raw output of the simulations, i.e., atomic positions. We demonstrate the method on crystal structure identification in Lennard-Jones (four different phases), water (eight different phases), and mesophase (six different phases) systems. The method achieves as high as 99.5% accuracy in crystal structure identification. The method is applicable to heterogeneous nucleation and it can even predict the crystal phases of atoms near external interfaces. We demonstrate the versatility of our approach by using our method to identify surface hydrophobicity based solely upon positions and orientations of surrounding water molecules. Our results suggest the approach will be broadly applicable to many types of local structure in simulations.
- This article is part of the themed collection: 2019 Chemical Science HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

