
How machine learning can assist the interpretation of ab initio molecular dynamics simulations and conceptual understanding of chemistry†

Abstract
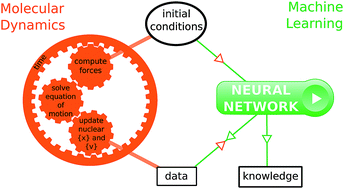
Molecular dynamics simulations are often key to the understanding of the mechanism, rate and yield of chemical reactions. One current challenge is the in-depth analysis of the large amount of data produced by the simulations, in order to produce valuable insight and general trends. In the present study, we propose to employ recent machine learning analysis tools to extract relevant information from simulation data without a priori knowledge on chemical reactions. This is demonstrated by training machine learning models to predict directly a specific outcome quantity of ab initio molecular dynamics simulations – the timescale of the decomposition of 1,2-dioxetane. The machine learning models accurately reproduce the dissociation time of the compound. Keeping the aim of gaining physical insight, it is demonstrated that, in order to make accurate predictions, the models evidence empirical rules that are, today, part of the common chemical knowledge. This opens the way for conceptual breakthroughs in chemistry where machine analysis would provide a source of inspiration to humans.
- This article is part of the themed collections: Most popular 2019-2020 physical and theoretical chemistry articles, Accelerating Chemistry Symposium Collection and 2019 Chemical Science HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

