
Hydrogen bond assisted photoinduced intramolecular electron transfer and proton coupled electron transfer in an ultrafast time domain using a ruthenium-anthraquinone dyad†

Abstract
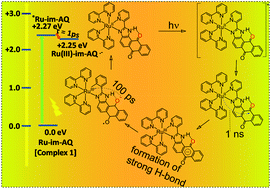
Quinones play a significant role as primary electron acceptors in the natural photosynthetic system of photosystem II, and their reduction is known to be facilitated by hydrogen-bond donors or protonation. In this study, a ruthenium(II) polypyridyl complex 1 coupled to an anthraquinone (AQ) functionality through a rigid imidazole (Im) spacer has been synthesized to examine the effect of H-bonding on both the thermal and photoinduced electron transfer reactions. The anthraquinone moiety of complex 1 is fused to a benzi-imidazole system bearing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OAQ⋯HNIm based H-bonding at one side of the anthraquinone moiety so that intramolecular hydrogen bonding from the imidazole group to the nearby quinone carbonyl can occur. The hydrogen bond formation involving the semiquinone radical anion produced through the photoinduced reduction process in Ru–im–AQ and the imidazole proton in complex 1 resulted in a significant positive shift of one electron reduction potential of complex 1. The kinetics for the formation of the charge-separated states was explored by using femtosecond transient absorption spectroscopy. Hydrogen bonding between water and the reduced anthraquinone accounted for thermodynamic and kinetic stabilization of these charge-separated states. An attempt has been made to assess the relative importance of the driving force and solvent polarity, in the rates of photoinduced electron transfer in complex 1. The 490 nm transient absorption band of anthraquinone radical anions (AQ˙−) and a broad absorption in the 580–750 nm region having maxima at ∼690 nm have been observed and this is attributed to the generation of a transient Ru3+-species of the corresponding complex 1. Addition of water entails an acceleration of electron transfer rates by a factor of 3.33. The system investigated may serve as a model for the mechanistic diversity of PCET reactions in general with water as a primary proton donor. Furthermore, our studies are relevant for understanding proton-coupled electron transfer (PCET) reactivity of electronically excited states at a fundamental level because changes in hydrogen-bonding strength accompanying changes in redox states may be regarded as a variant form of PCET.
OAQ⋯HNIm based H-bonding at one side of the anthraquinone moiety so that intramolecular hydrogen bonding from the imidazole group to the nearby quinone carbonyl can occur. The hydrogen bond formation involving the semiquinone radical anion produced through the photoinduced reduction process in Ru–im–AQ and the imidazole proton in complex 1 resulted in a significant positive shift of one electron reduction potential of complex 1. The kinetics for the formation of the charge-separated states was explored by using femtosecond transient absorption spectroscopy. Hydrogen bonding between water and the reduced anthraquinone accounted for thermodynamic and kinetic stabilization of these charge-separated states. An attempt has been made to assess the relative importance of the driving force and solvent polarity, in the rates of photoinduced electron transfer in complex 1. The 490 nm transient absorption band of anthraquinone radical anions (AQ˙−) and a broad absorption in the 580–750 nm region having maxima at ∼690 nm have been observed and this is attributed to the generation of a transient Ru3+-species of the corresponding complex 1. Addition of water entails an acceleration of electron transfer rates by a factor of 3.33. The system investigated may serve as a model for the mechanistic diversity of PCET reactions in general with water as a primary proton donor. Furthermore, our studies are relevant for understanding proton-coupled electron transfer (PCET) reactivity of electronically excited states at a fundamental level because changes in hydrogen-bonding strength accompanying changes in redox states may be regarded as a variant form of PCET.
 Please wait while we load your content...
Please wait while we load your content...