
Pipecolic esters as minimized templates for proteasome inhibition†

Abstract
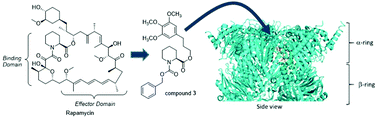
Allosteric regulators of clinically important enzymes are gaining popularity as alternatives to competitive inhibitors. This is also the case for the proteasome, a major intracellular protease and a target of anti-cancer drugs. All clinically used proteasome inhibitors bind to the active sites in catalytic chamber and display a competitive mechanism. Unfortunately, inevitable resistance associated with this type of inhibition drives the search for non-competitive agents. The multisubunit and multicatalytic “proteolytic machine” such as the proteasome is occasionally found to be affected by agents with other primary targets. For example the immunosuppressive agent rapamycin has been shown to allosterically inhibit the proteasome albeit at levels far higher than its mTOR related efficacy. As part of an ongoing program to search for novel proteasome-targeting pharmacophores, we identified the binding domain of rapamycin as required for proteasome inhibition even without the macrocyclic context of the parent compound. By subsequent structure–activity relationship studies, we generated a pipecolic ester derivative compound 3 representing a new class of proteasome inhibitors. Compound 3 affects the core proteasome activities and proliferation of cancer cells with low micromolar/high nanomolar efficacy. Molecular modeling, atomic force microscopy imaging and biochemical data suggest that compound 3 binds into one of intersubunit pockets in the proteasomal α ring and destabilizes the α face and the gate. The α face is used as a docking area for proteasome-regulating protein modules and the gate is critical for controlling access to the catalytic chamber. Thus, the pipecolic ester template elicits a new and attractive mechanism for proteasome inhibition distinct from classical competitive drugs.
- This article is part of the themed collection: Chemical Biology in OBC
 Please wait while we load your content...
Please wait while we load your content...

