
Targeting the hydrophobic channel of NNIBP: discovery of novel 1,2,3-triazole-derived diarylpyrimidines as novel HIV-1 NNRTIs with high potency against wild-type and K103N mutant virus

Abstract
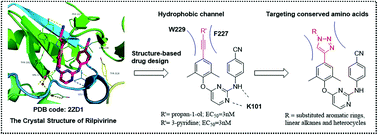
Enlightened by our previous efforts to modify diarylpyrimidines as HIV-1 non-nucleoside reverse transcriptase (RT) inhibitors (NNRTIs) and the reported crystallographic studies, we designed and synthesized novel 1,2,3-triazole-derived diarylpyrimidine derivatives via the CuAAC “click reaction”, to make additional interactions with the hydrophobic channel in the NNRTI binding pocket. The newly synthesized compounds were evaluated for anti-HIV potency in MT-4 cells. All the compounds showed favorable activity against the wild-type HIV-1 strain with an EC50 of 0.013–5.62 μM. Interestingly, some compounds displayed remarkable potency in inhibiting K103N mutant virus, a key drug-resistant mutant to NNRTIs. Among them, meta-methylbenzoate (ZL2, EC50(IIIB) = 0.020 μM, EC50(K103N) = 0.043 μM, CC50 > 241.52 μM), para-methylbenzoate (ZL3, EC50(IIIB) = 0.013 μM, EC50 (K103N) = 0.022 μM, CC50 > 241.52 μM) and para-phenol (ZL7, EC50(IIIB) = 0.014 μM, EC50 (K103N) = 0.054 μM, CC50 = 2.1 μM) derivatives are the three most promising compounds which are superior to the first-line antiretroviral drug efavirenz (EC50(IIIB) = 0.003 μM, EC50 (K103N) = 0.11 μM, CC50 > 6.34 μM) against the K103N mutant strain. More encouragingly, ZL2 and ZL3 exhibited much lower cytotoxicity and a high selection index of >10 000 compared with all the control drugs (AZT, 3TC, NVP, EFV, and ETV). The detailed structure–activity relationship (SAR), enzymatic inhibitory activity and docking study of the representative compounds are also discussed. Furthermore, the preliminary physicochemical properties and the early metabolic stability of representative compounds were examined to evaluate their drug-like properties.
- This article is part of the themed collection: Chemical Biology in OBC
 Please wait while we load your content...
Please wait while we load your content...

