
Titanium-decorated boron nitride nanotubes for hydrogen storage: a multiscale theoretical investigation†

Abstract
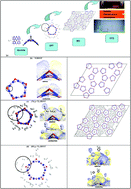
A multiscale computational technique from available quantum mechanical and Molecular Dynamics (MD) codes to user-subroutine computational fluid dynamics (CFD) files was applied to the H2 adsorption of Ti-decorated (10,0) single-walled BN nanotubes (BNNTs) with B–N defects. According to density functional theory (DFT), the Ti atom adsorbed in the defect protrudes outward from the surface and does not agglomerate. The Ti/BNNT system possesses excellent affinity towards H2 molecules with thermodynamically favorable binding energies. Up to seven H2 molecules can partially attach near Ti in quasi-molecular form due to the cationic functionalized Ti and heteropolar B–N bonds at the surface. MD simulation further reveals that the seven H2 molecules are indeed physisorbed in two distinct regions near to (at ∼2 Å) and far from (at ∼4 Å) the Ti atom. The thermal properties obtained from MD simulations were utilized in the CFD code to quantify thermal energy transfer. According to CFD, the equilibrium pressure distribution for a type III H2 cylinder is initially low within its locality. The rate of change of pressure increase is quite steep as it rises due to a sudden increase in temperature upon uptake and it eventually slows down when the local temperature reaches its saturation value.
 Please wait while we load your content...
Please wait while we load your content...

