
Remote control of charge transport and chiral induction along a DNA-metallohelicate†

Abstract
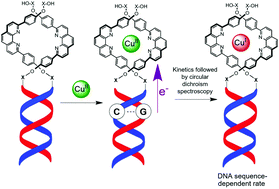
Herein we present a new strategy to achieve chiral induction and redox switching along the backbone of metallohelicate architectures, wherein a DNA duplex directs the handedness and charge transport properties of a metal–organic assembly more than 60 bonds away (a distance of >10 nm). The quantitative and site-specific binding of copper(I) ions to DNA-templated coordination sites imparts enhanced thermodynamic stability to the assembly, while the DNA duplex transfers its natural right-handed helicity to the proximal and distal metal centers of the helicates. When copper(II) ions are employed instead of copper(I) ions, spontaneous DNA-mediated reduction occurs, which we propose is followed by a slower change in coordination environment (from pentacoordinate CuII to tetrahedral CuI) to generate copper(I) helicates. We demonstrate that the reduction of the adjacent and distal bis-phenanthroline sites is dependent on their proximity to DNA guanine bases (which act as the electron source). The kinetics of helical charge transport can thus be tuned based on guanine-CuII separation, resulting in a sequence- and distance-dependent redox switch that transfers electronic information from DNA to multiple linearly-arranged metal centers.
 Please wait while we load your content...
Please wait while we load your content...

