
Nature of the E⋯E′ interactions (E, E′ = O, S, Se, and Te) at naphthalene 1,8-positions with fine details of the structures: experimental and theoretical investigations†

Abstract
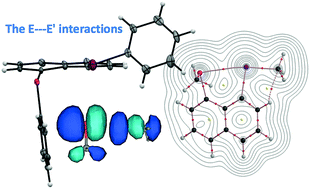
The intrinsic dynamic and static natures of the E⋯E′ interactions at the 1,8-positions of 1-(MeE)–8-(MeE′)C10H6 [1a–1f (E ≠ E′) and 1g–1j (E = E′)] were elucidated with QTAIM-DFA, after structural determination of 1-(PhE)–8-(PhE′)C10H6 (3a–3f), where (E, E′: x) = (O, S: a), (O, Se: b), (O, Te: c), (S, Se: d), (S, Te: e), (Se, Te: f), (O, O: g), (S, S: h), (Se, Se: i) and (Te, Te: j) (χE ≥ χE′). While the AB structures are confirmed for 3a, 3b and 3d–3f, which consist of the np(E)⋯σ*(E′–CPh) interactions, the structure was BB for 3c, where the E–CR/E′–CR (R = Ph) bond is perpendicular to the naphthyl plane in A and it is placed on the plane in B. While the AB structures are determined by the p(E)–π(Ph) conjugations, the BB structure is by the crystal packing effect. The BA structure with np(E′)⋯σ*(E–CPh) was not detected. While the nature of a typical hydrogen bond with covalency was predicted for BB, AA and BA, with the CT-MC (molecular complex formation through charge transfer) nature for AB in 1e and 1f (R = Me for E–CR/E′–CR), the CT-MC nature was predicted for all conformers of 1j, for example. NBO analysis for 1a–1f revealed that the acceptor orbitals contribute much more than the donor orbitals and the order is σ*(O–CMe: <0.5 kcal mol−1) ≪ σ*(S–CMe: ≈5 kcal mol−1) ≪ σ*(Se–CMe: ≈10 kcal mol−1) ≪ σ*(Te–CMe: ≈16 kcal mol−1) for np(S), np(Se) and np(Te). The E(2) values proportionally correlate to Cii−1 for AB and/or BA of 1a–1j, if analyzed separately for σ*(S–C), σ*(Se–C) and σ*(Te–C).
- This article is part of the themed collection: Selenium & Tellurium chemistry at the beginning of the 3rd millennium: a celebration of ICCST
 Please wait while we load your content...
Please wait while we load your content...

