
Anthranilic amide and imidazobenzothiadiazole compounds disrupt Mycobacterium tuberculosis membrane potential†
 *a
*a
Abstract
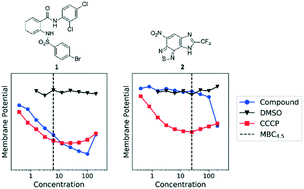
A family of compounds typified by an anthranilic amide 1 was identified from a whole-cell screening effort targeted at identifying compounds that disrupt pH homeostasis in Mycobacterium tuberculosis. 1 demonstrated bactericidal activity against non-replicating M. tuberculosis in pH 4.5 buffer (MBC4.5 = 6.3 μM). Exploration of the structure–activity relations failed to simplify the scaffold. The antitubercular activity proved dependent on the lipophilicity and planarity of the molecule and directly correlated with mammalian cytotoxicity. Further studies revealed a pH-dependent correlation between the family's disruption of M. tuberculosis membrane potential and antitubercular activity, with active compounds causing a drop in membrane potential at concentrations below their MBC4.5. A second compound family, identified in the same screening effort and typified by imidazo(4,5-e)(2,1,3)benzothiadiazole 2, provided a contrasting profile. As with 1, structure–activity profiling of 2 (MBC4.5 = 25 μM) failed to minimize the initial scaffold, mammalian cytotoxicity was observed for a majority of the active compounds, and many of the active compounds disrupted M. tuberculosis membrane potential. However, unlike the anthranilic amide compounds, the benzothiadiazole compounds disrupted M. tuberculosis membrane potential primarily at concentrations above the MBC4.5 in a pH-independent fashion. These differences suggest an alternative mechanism of action for the benzothiadiazole compounds. As a result, while the cytotoxicity of the anthranilic amides limits their utility to tool compounds, benzothiadiazole 2 presents an attractive target for more focused SAR exploration.
- This article is part of the themed collections: Antimicrobial Resistance and Molecular Pharmacology
 Please wait while we load your content...
Please wait while we load your content...