
Molecular mechanisms for thermal degradation of CO2-loaded aqueous monoethanolamine solution: a first-principles study†
 *ab
*ab
Abstract
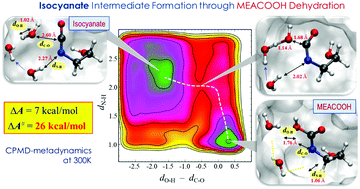
Thermal degradation of aqueous monoethanolamine (MEA), a benchmark solvent, in CO2 capture processes still remains a challenge. Here, we present molecular mechanisms underlying thermal degradation of MEA based on ab initio molecular dynamics simulations coupled with metadynamics sampling. Isocyanate formation via dehydration of carbamic acid (MEACOOH) is predicted to be highly probable and more kinetically favorable than the competing cyclization–dehydration reaction to 2-oxazolidinone (OZD), albeit not substantially. Isocyanate may undergo cyclization to form OZD, which is found to be more facile in aqueous MEA solution than reaction with MEA to form urea, although the latter is thermodynamically more favorable than the former. Our simulations also clearly demonstrate that OZD is a long-lived intermediate that plays a key role in MEA thermal degradation to experimentally observed products. Overall, this work highlights the importance of entropic contributions associated with the local structure and dynamics of solvent molecules around the intermediates, which cannot be solely explained by thermodynamics, in predicting the mechanism and kinetics of thermal degradation of CO2-loaded aqueous amine solutions.
 Please wait while we load your content...
Please wait while we load your content...

