
Structure and electronic properties of rare earth DOBDC metal–organic-frameworks†

Abstract
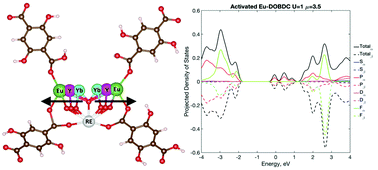
Here, we apply density functional theory (DFT) to investigate rare-earth metal organic frameworks (RE-MOFs), RE12(μ3-OH)16(C8O6H4)8(C8O6H5)4 (RE = Y, Eu, Tb, Yb), and characterize the level of theory needed to accurately predict structural and electronic properties in MOF materials with 4f-electrons. A two-step calculation approach of geometry optimization with spin-restricted DFT and large core potential (LCPs), and detailed electronic structures with spin-unrestricted DFT with a full valence potential + Hubbard U correction is investigated. Spin-restricted DFT with LCPs resulted in good agreement between experimental lattice parameters and optimized geometries, while a full valence potential is necessary for accurate representation of the electronic structure. The electronic structure of Eu-DOBDC MOF indicated a strong dependence on the treatment of highly localized 4f-electrons and spin polarization, as well as variation within a range of Hubbard corrections (U = 1–9 eV). For Hubbard corrected spin-unrestricted calculations, a U value of 1–4 eV maintains the non-metallic character of the band gap with slight deviations in f-orbital energetics. When compared with experimentally reported results, the importance of the full valence calculation and the Hubbard correction in correctly predicting the electronic structure is highlighted.
 Please wait while we load your content...
Please wait while we load your content...

