
Excited-state non-radiative decay in stilbenoid compounds: an ab initio quantum-chemistry study on size and substituent effects†

Abstract
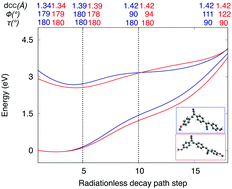
In the framework of optoelectronic luminescent materials, non-radiative decay mechanisms are relevant to interpret efficiency losses. These radiationless processes are herein studied theoretically for a series of stilbenoid derivatives, including distyrylbenzene (DSB) and cyano-substituted distyrylbenzene (DCS) molecules in vacuo. Given the difficulties of excited-state reaction path determinations, a simplified computational strategy is defined based on the exploration of the potential energy surfaces (PES) along the elongation, twisting, and pyramidalization of the vinyl bonds. For such exploration, density functional theory (DFT), time-dependent (TD)DFT, and complete-active-space self-consistent field/complete-active-space second-order perturbation theory (CASSCF/CASPT2) are combined. The strategy is firstly benchmarked for ethene, styrene, and stilbene; next it is applied to DSB and representative DCS molecules. Two energy descriptors are derived from the approximated PES, the Franck–Condon energy and the energy gap at the elongated, twisted, and pyramidalized structures. These energy descriptors correlate fairly well with the non-radiative decay rates, which validates our computational strategy. Ultimately, this strategy may be applied to predict the luminescence behavior in related compounds.
 Please wait while we load your content...
Please wait while we load your content...

