
NMR shifts in aluminosilicate glasses via machine learning†
 *a
Thibault
Charpentier
*a
*a
Thibault
Charpentier
*a
Abstract
Machine learning (ML) approaches are investigated for the prediction of nuclear magnetic resonance (NMR) parameters in aluminosilicate glasses, for which NMR has proven to be a cutting-edge method over the last decade. DFT computations have emerged as a new dimension for complementing these NMR methods although suffering from severe limitations in terms of size, time and computational resources consumption. While previous approaches tend to use DFT-GIPAW calculations for the prediction of NMR parameters in glassy systems, we propose to employ ML methods, characterized by a speed similar to that of classical molecular dynamics while the accuracy of ab initio methods can be reached. We design ML procedures to predict the isotropic magnetic shielding (σiso) for different multicomponent relevant glass compositions. The ML predictions of σiso deviate from DFT-GIPAW calculations, when including relaxed and room-temperature structures, by 0.7 ppm for 29Si (1.0% of the total span of the calculated 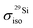 ) and 1.5 ppm for 17O (1.9%) in SiO2 glasses, 1.4 ppm for 23Na (1.5%) in Na2O–SiO2 and 1.5 ppm for 27Al (2.1%) in Al2O3–Na2O–SiO2 systems. We compare the performances obtained for a set of three descriptors suitable for encoding atomic local environments information (atom-centered representations) together with seven popular ML algorithms with a focus on the simple (but robust) linear ridge regression (LRR) and the popular smooth overlap of atomic positions (SOAP) descriptor.
) and 1.5 ppm for 17O (1.9%) in SiO2 glasses, 1.4 ppm for 23Na (1.5%) in Na2O–SiO2 and 1.5 ppm for 27Al (2.1%) in Al2O3–Na2O–SiO2 systems. We compare the performances obtained for a set of three descriptors suitable for encoding atomic local environments information (atom-centered representations) together with seven popular ML algorithms with a focus on the simple (but robust) linear ridge regression (LRR) and the popular smooth overlap of atomic positions (SOAP) descriptor.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

