
Cooperativity and coverage dependent molecular desorption in self-assembled monolayers: computational case study with coronene on Au(111) and HOPG†

Abstract
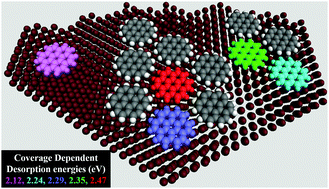
One of the common practices in the literature of molecular desorption is the comparison of theoretically (mostly using DFT) calculated single molecule adsorption energies with experimental desorption energies from studies like temperature programmed desorption (TPD) etc. Comparisons like those do not consider that the experimental desorption energies are obtained via ensemble techniques while theoretical values are calculated at the single molecule level. Theoretical values are generally based upon desorption of a single molecule from a clean surface, or upon desorption of an entire monolayer. On the other hand, coverage dependent molecule–molecule interactions add to and modify molecule–substrate interactions that contribute to the experimentally determined desorption energies. In this work, we explore the suitability of an additive nearest neighbor model for determining general coverage dependent single molecule desorption energies in non-covalent self-assembled monolayers (SAMs). These coverage dependent values serve as essential input to any model attempting to reproduce coverage dependent desorption or for understanding the time dependent desorption from a partially covered surface. This method is tested using a case study of coronene adsorbed on Au(111) and HOPG substrates with periodic DFT calculations. Calculations show that coronene exhibits coverage and substrate dependence in molecular desorption. We found that intermolecular contact energies in the coronene monolayer are not strongly influenced by the HOPG substrate, while coronene desorption on Au(111) exhibits strong cooperativity where the additive model fails.
 Please wait while we load your content...
Please wait while we load your content...

