
Molecular dimers of methane clathrates: ab initio potential energy surfaces and variational vibrational states†

Abstract
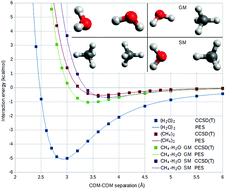
Motivated by the energetic and environmental relevance of methane clathrates, highly accurate ab initio potential energy surfaces (PESs) have been developed for the three possible dimers of the methane and water molecules: (H2O)2, CH4·H2O, and (CH4)2. While only a single monomer geometry was used for each monomer in the ab initio calculations, the PES parameterization makes it possible to produce distinct surfaces for all isotopologues within the rigid-monomer approximation. The PESs were fitted to computations at the frozen-core coupled-cluster level with single, double, and non-iterative triple excitations, employing basis sets of augmented triple- and quadruple-zeta quality plus bond functions, followed by extrapolations to the complete basis set limit. The long-range parts of the PESs are computed using the asymptotic version of symmetry-adapted perturbation theory based on a density-functional description of the monomers. All PESs are polarizable, i.e., in cluster or condensed-phase applications they approximate many-body effects by the induced dipole polarization model. The PESs were developed in a fully automated procedure applying the autoPES method, which is used for the first time to generate near-spectroscopic quality surfaces. The stationary points (SPs) on the PESs have been determined and compared with literature data. For CH4·H2O, previously unknown SPs have been identified and the first detailed study of the (CH4)2 potential energy landscape has been carried out. The PESs were used in variational quantum nuclear motion computations. For the water dimer, the resulting vibrational transitions are in excellent agreement with available high-resolution spectroscopic data. For (CH4)2, the intermonomer vibrational states are reported for the first time.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

