
Toward fast and accurate ab initio calculation of magnetic exchange in polynuclear lanthanide complexes

Abstract
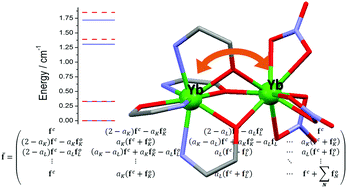
Ab initio calculations of the magnetic exchange in polynuclear lanthanide complexes are very challenging and often not feasible, due to large active spaces, the large number of required states or the necessity to include dynamical correlation into the calculations. We present an approach which allows for the computationally efficient calculation of exchange splittings in polynuclear lanthanide complexes including dynamical correlation. This is achieved by extending the local-density-fitted configuration-averaged Hartree–Fock (LDF-CAHF) method to systems with more than one group of open-shell orbitals (e.g. at different metal atoms) and combining it with linear-scaling many-state pair-natural-orbital complete active space perturbation theory of second order (PNO-CASPT2). In order to assess the performance of the method, we apply it to the asymmetric dinuclear complex [hqH2][Yb2(hq)4(NO3)3]·MeOH (hqH = 8-hydroxyquinoline).
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

