
Excited states and excitonic interactions in prototypic polycyclic aromatic hydrocarbon dimers as models for graphitic interactions in carbon dots†
 a
Dana
Nachtigallová,
*bc
Adélia J. A.
Aquino,
a
Francisco B. C.
Machado
d
and
Hans
Lischka
*a
a
Dana
Nachtigallová,
*bc
Adélia J. A.
Aquino,
a
Francisco B. C.
Machado
d
and
Hans
Lischka
*a
Abstract
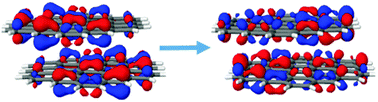
The study of electronically excited states of stacked polycyclic aromatic hydrocarbons (PAHs) is of great interest due to promising applications of these compounds as luminescent carbon nanomaterials such as graphene quantum dots (GQDs) and carbon dots (CDs). In this study, the excited states and excitonic interactions are described in detail based on four CD model dimer systems of pyrene, coronene, circum-1-pyrene and circum-1-coronene. Two multi-reference methods, DFT/MRCI and SC-NEVPT2, and two single-reference methods, ADC(2) and CAM-B3LYP, have been used for excited state calculations. The DFT/MRCI method has been used as a benchmark method to evaluate the performance of the other ones. All methods produce useful lists of excited states. However, an overestimation of excitation energies and an inverted ordering of states, especially concerning the bright HOMO–LUMO excitation, are observed. In the pyrene-based systems, the first bright state appears among the first four states, whereas the number of dark states is significantly larger for the coronene-based systems. Fluorescence emission properties are addressed by means of geometry optimization in the S1 state. The inter sheet distances for the S1 state decrease in comparison to the corresponding ground-state values. These reductions are largest for the pyrene dimer and decrease significantly for the larger dimers. Several minima have been determined on the S1 energy surface for most of the dimers. The largest variability in emission energies is found for the pyrene dimer, whereas in the other cases a more regular behavior of the emission spectra is observed.
- This article is part of the themed collection: Bunsentagung 2019: Functional Materials
 Please wait while we load your content...
Please wait while we load your content...

