
Computational study of the carbonyl–ene reaction between formaldehyde and propylene encapsulated in coordinatively unsaturated metal–organic frameworks M3(btc)2 (M = Fe, Co, Ni, Cu and Zn)†
 *a
*a
Abstract
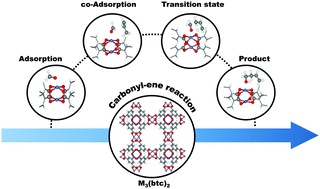
The carbonyl–ene reaction between encapsulated formaldehyde and propylene over the coordinatively unsaturated metal–organic frameworks M3(btc)2 (M = Fe, Co, Ni, Cu and Zn) has been investigated by means of density functional calculations. Zn3(btc)2 adsorbs formaldehyde strongest due to electron delocalization between Zn and the oxygen atom of the reactant molecule. The reaction is proposed to proceed in a single step involving proton transfer and carbon–carbon bond formation. We find the relative catalytic activity to be Zn3(btc)2 > Fe3(btc)2 ≥ Co3(btc)2 > Ni3(btc)2 > Cu3(btc)2, based on activation energy and turnover frequencies (TOF). The low activation energy for Zn3(btc)2 compared to the others can be explained by the delocalization of electron density between the carbonyl bond and the catalyst active sites, leading to a more stable transition state. The five MOFs are used to propose a descriptor for the relationship between activation energy on one side and electronic properties or adsorption energies on the other side in order to allow a quick screening of other catalytic materials for this reaction.
 Please wait while we load your content...
Please wait while we load your content...

