
Theoretical X-ray absorption spectroscopy database analysis for oxidised 2D carbon nanomaterials†

Abstract
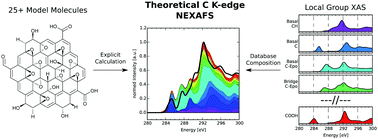
In this work we provide a proof of principle for a theoretical methodology to identify functionalisation patterns in oxidised carbon 2D nanomaterials. The methodology is based on calculating a large number of X-ray absorption spectra of individually excited carbon atoms in different chemical environments using density functional theory. Since each resulting spectrum gives a fingerprint of the local electronic structure surrounding the excited atom, we may relate each spectrum to the functionalisation pattern of that excited atom up to a desired neighbourhood radius. These functionalisation pattern-specific spectra are collected in a database, that allows fast composition of X-ray absorption spectra for arbitrary structures in density functional theory quality. Finally, we present an exemplary application of the database approach to estimate the relative amount of functional groups in two different experimental samples of carbon nanomaterials.
 Please wait while we load your content...
Please wait while we load your content...

