
Towards high-level theoretical studies of large biodiesel molecules: an ONIOM/RRKM/Master-equation approach to the isomerization and dissociation kinetics of methyl decanoate radicals†
 *b
*b
Abstract
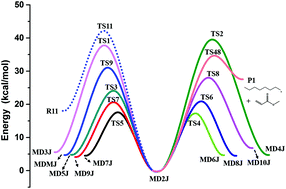
The isomerization and dissociation reactions of methyl decanoate (MD) radicals were theoretically investigated by using high-level theoretical calculations based on a two-layer ONIOM method, employing the QCISD(T)/CBS method for the high layer and the M06-2X/6-311++G(d,p) method for the low layer. Temperature- and pressure-dependent rate coefficients for the involved reactions were computed by using the transition state theory and the Rice–Ramsperger–Kassel–Marcus/Master-equation method. The structure–reactivity relationships were explored for the complicated multiple-well interconnected system of ten isomeric MD radicals. Comparative studies of methyl butanoate (MB) and MD were also performed systematically. Results show that the isomerization reactions are appreciably responsible for the population distribution of MD radicals at low and intermediate temperatures, while the β-scission reactions are dominant at higher temperatures. Although the rate constants of MB specific to methyl esters are close to those of MD in certain temperature ranges, MB is unable to simulate most of the dissociation reactions due to its short aliphatic chain. Significant differences of rate constants for isomerization reactions were observed between the calculated results and the literature data, which were estimated by analogy to alkane systems, but the rate constants of β-scissions show generally good agreement between theory and experiment. The current work extends kinetic data for isomerization and dissociation reactions of MD radicals, and it serves as a reference for the studies of detailed combustion chemistry of practical biodiesels.
 Please wait while we load your content...
Please wait while we load your content...

