
Design principles for the energy level tuning in donor/acceptor conjugated polymers
 †a
John
Kieffer
*a
†a
John
Kieffer
*a
Abstract
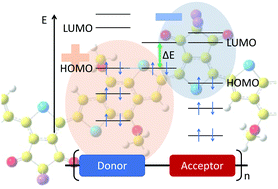
To identify reliable molecular design principles for energy level tuning in donor/acceptor conjugated polymers (CPs), we studied the governing factors by means of ab initio calculations based on density-functional theory (DFT). We investigated a series of CPs in which we independently and systematically varied the electron withdrawing power of the acceptor unit and the electron donating power of the donor unit, while maintaining the same conjugated chain conformation. We observed that the introduction of a stronger acceptor unit, while keeping the same donor unit in the CP, lowers the LUMO level, but leaves the HOMO level almost unchanged. Conversely, enhancing the strength of the donor unit for the same acceptor unit raises the HOMO level, while maintaining the LUMO level. We identified strong correlations between the frontier orbital energy levels and the degree of orbital localization, which depends on the electron donating or withdrawing power of the molecular groups carrying the orbitals. Moreover, the HOMO/LUMO gap of the CP is directly proportional to the charge transfer between donating and accepting units, which provides a robust design criterion for CPs.
 Please wait while we load your content...
Please wait while we load your content...

