
Exploratory machine-learned theoretical chemical shifts can closely predict metabolic mixture signals†
 ab
Jun
Kikuchi
*abd
ab
Jun
Kikuchi
*abd
Abstract
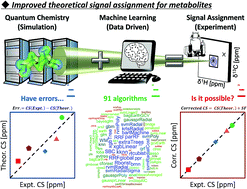
Various chemical shift predictive methodologies have been studied and developed, but there remains the problem of prediction accuracy. Assigning the NMR signals of metabolic mixtures requires high predictive performance owing to the complexity of the signals. Here we propose a new predictive tool that combines quantum chemistry and machine learning. A scaling factor as the objective variable to correct the errors of 2355 theoretical chemical shifts was optimized by exploring 91 machine learning algorithms and using the partial structure of 150 compounds as explanatory variables. The optimal predictive model gave RMSDs between experimental and predicted chemical shifts of 0.2177 ppm for δ1H and 3.3261 ppm for δ13C in the test data; thus, better accuracy was achieved compared with existing empirical and quantum chemical methods. The utility of the predictive model was demonstrated by applying it to assignments of experimental NMR signals of a complex metabolic mixture.
 Please wait while we load your content...
Please wait while we load your content...

