
Cobalt-catalysed alkene hydrogenation: a metallacycle can explain the hydroxyl activating effect and the diastereoselectivity†

Abstract
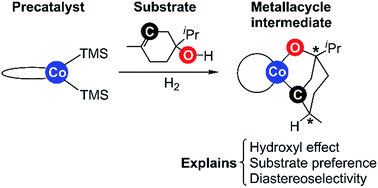
Bis(phosphine)cobalt dialkyl complexes have been reported to be highly active in the hydrogenation of tri-substituted alkenes bearing hydroxyl substituents. Alkene substrates containing ether, ester, or ketone substituents show minimal reactivity, indicating an activating effect of the hydroxyl group. The mechanistic details of bis(phosphine)cobalt-catalysed hydrogenation were recently evaluated computationally (X. Ma, M. Lei, J. Org. Chem. 2017, 82, 2703–2712) and a Co(0)–Co(II) redox mechanism was proposed. However, the activating effect of the hydroxyl substituent and the accompanying high diastereoselectivity were not studied. Here we report a computational study rationalizing the role of the hydroxyl group through a key metallacycle species. The metallacycle is part of a non-redox catalytic pathway proceeding through Co(II) intermediates throughout. The preference for alcohol over ether substrates and the high diastereoselectivity of terpinen-4-ol hydrogenation are correctly predicted in computations adopting the new pathway, whereas the alternative redox mechanism predicts ethers rather than alcohols to be more reactive substrates. Additional experimental evidence supports the role of the hydroxyl group in the metallacycle mechanism. Our work highlights the importance of employing known substrate preferences and stereoselectivities to test the validity of computationally proposed reaction pathways.
 Please wait while we load your content...
Please wait while we load your content...

