
NMR-based investigations of acyl-functionalized piperazines concerning their conformational behavior in solution†

Abstract
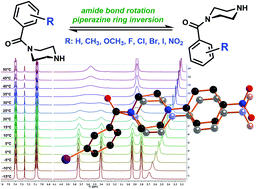
Selected N-benzoylated piperazine compounds were synthesized to study their conformational behavior using temperature-dependent 1H NMR spectroscopy. All investigated piperazines occur as conformers at room temperature resulting from the restricted rotation of the partial amide double bond. In the case of selected mono-N-benzoylated and unsymmetrically N,N′-substituted derivatives, the appearance of the 1H NMR spectrum was further shaped by the limited interconversion of the piperazine chair conformations. Therefore, two different coalescence points TC were determined and their resulting activation energy barriers ΔG‡ were calculated to be between 56 and 80 kJ mol−1. In most of the cases, TC and ΔG‡ for the amide site appeared to be higher than the corresponding values for the ring inversion. The influences of substituents on rotational and inversion barriers were analyzed by correlation to Hammett constants. The obtained results are discussed and interpreted in the context of literature data. An additional aryl substituent connected at the amine site led to reduced rotational and inversion barriers compared to the free secondary amine. To support and evidence the findings from the NMR analyses, single crystals of selected piperazines were obtained and XRD analyses were performed. To underline the results, two potential TGase 2 inhibitors were investigated showing energy barriers with similar values.
 Please wait while we load your content...
Please wait while we load your content...

