
Computational investigation of the control of the thermodynamics and microkinetics of the reductive amination reaction by solvent coordination and a co-catalyst†

Abstract
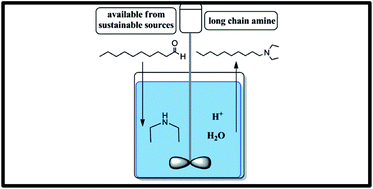
Amines are among the most important and frequently used chemical compounds due to their biological activity and a wide range of applications in industry. Reductive amination reactions are an efficient and facile route to synthesize long chain amines from sustainable sources by using a different available aldehydes and ketones, and a large variety of amines including primary, secondary and tertiary forms. The pathway of the reaction process is critically dependent on reaction parameters such as the pH of the reaction medium, choice of solvent (explicitly coordinating solvent) and process conditions. These parameters are affecting the reaction performance and the selectivity but are still not fully rationalized. Here, we investigate the microkinetics and thermodynamics of the individual steps of the reductive amination reaction by exploring the systems' parameters. Explicit water coordination to the aldehyde leads to a stepwise rather than concerted nucleophilic addition with a lower activation barrier by 6–10 kcal mol−1. At low pH, the pathway is changed by a direct protonation of the amine substrate. This protonation does not strongly affect the kinetics of the reaction, but the thermodynamic equilibria. The presence of an acid as a co-catalyst leads to the formation of an iminium intermediate and this drives the reaction forward. Thus, the presence of an acid as a co-catalyst clearly renders this pathway the thermodynamically preferred one. Consequently, altering the reaction parameters does not only influence the reaction kinetics, but also the thermodynamic profile of the pathways in all cases. Further understanding of the reaction dynamics is essential to develop a microkinetic model of the reaction to then control and engineer the process in order to rationally design routes to tailor-made products.
 Please wait while we load your content...
Please wait while we load your content...

