
Mechanistic insights into the different chemoselectivities of Rh2(ii)-catalyzed ring expansion of cyclobutanol-substituted aryl azides and C–H bond amination of cyclopentanol-substituted aryl azides: a DFT study†

Abstract
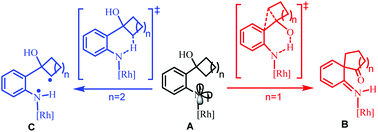
The mechanism of Rh2(II)-catalyzed ring expansion of ortho-cyclobutanol-substituted aryl azides to access N-heterocycles was investigated by DFT calculations. After the generation of the key Rh2(II)-N-arylnitrene intermediate (A), computational results suggest that it is more feasible to undergo proton transfer from the O–H group of the ortho-cyclobutanol moiety to the nitrene site in the singlet state, along with four-membered ring expansion via [1,2] migration in a concerted manner, to form the key intermediate (B). Subsequently, the [1,3] migration step driven by rearomatization could follow to yield the final benzazepinone product. For the substrate, ortho-cyclopentanol-substituted aryl azide, H-atom abstraction (HAA) from the proximal sp3 C–H bond in the triplet state is more favorable than competitive HAA/proton transfer from the O–H group after the formation of the rhodium N-aryl nitrene intermediate. Subsequently, the C–H bond amination product, indoline, could be obtained via a radical rebound step. The origin of the different chemoselectivities for the ortho-cyclobutanol-substituted and ortho-cyclopentanol-substituted aryl azides was discussed.
 Please wait while we load your content...
Please wait while we load your content...

