
Human cytochrome P450 enzyme inhibition profile of three flavonoids isolated from Psoralea corylifolia: in silico predictions and experimental validation
 *ab
*ab
Abstract
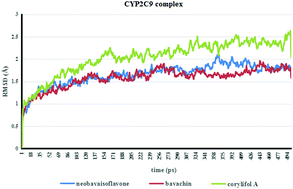
Cytochrome P450 enzyme (CYP)-associated metabolic studies in vitro have been considered cost-effective for predicting potential clinical drug/herb–drug interactions (DDI/HDI). There is an increasing use of computational approaches to investigate CYPs and their interactions with ligands. In silico studies will help us to significantly reduce the number of potential false positive hypotheses and decrease the running times of the experiments. They can also better predict and comprehend the molecular nature of binding interactions. Bavachin, neobavaisoflavone and corylifol A are the bioactive flavonoids of Psoralea corylifolia, which have been found to be associated with osteoarthritis treatment. In this study, molecular docking (CDOCKER) together with molecular dynamic simulation (MD) methods was used to predict the binding mode of these three flavonoids with CYP1A2/2C9/2C19/2D6/3A4/2E1. The IC50 shift and GSH-trapping experiments were conducted to validate the computational results as well as to identify the inhibition mechanism. The binding abilities predicted by CDOCKER and MD simulation with CYPs were in good agreement with the experimental data. Further inhibitory mechanism studies showed that bavachin and neobavaisoflavone might be mechanism-based inhibitors of CYP1A2 and CYP2C19. These results suggest that the flavonoids obtained from Psoralea corylifolia may participate in interactions with drugs that act as substrates for CYP2C9, CYP2C19 and CYP1A2, and the combination of CDOCKER and MD simulation may be a useful tool to identify potential CYP inhibitors from herbal medicines.
 Please wait while we load your content...
Please wait while we load your content...

