
Structure activity relationship of 2-arylalkynyl-adenine derivatives as human A3 adenosine receptor antagonists†

Abstract
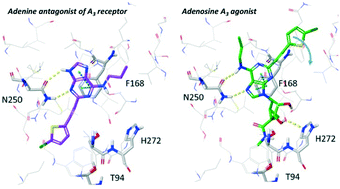
Recognition of nucleosides at adenosine receptors (ARs) is supported by multiple X-ray structures, but the structure of an adenine complex is unknown. We examined the selectivity of predicted A1AR and A3AR adenine antagonists that incorporated known agonist affinity-enhancing N6 and C2 substituents. Adenines with A1AR-favoring N6-alkyl, cycloalkyl and arylalkyl substitutions combined with an A3AR-favoring 2-((5-chlorothiophen-2-yl)ethynyl) group were human (h) A3AR-selective, e.g. MRS7497 17 (∼1000-fold over A1AR). In addition, binding selectivity over hA2AAR and hA2BAR and functional A3AR antagonism were demonstrated. 17 was subjected to computational docking and molecular dynamics simulation in a hA3AR homology model to predict interactions. The SAR of nucleoside AR agonists was not recapitulated in adenine AR antagonists, and modeling suggested an alternative, inverted binding mode with the key N2506.55 H-bonding to the adenine N3 and N9, instead of N6 and N7 as in adenosine agonists.
- This article is part of the themed collection: Molecular Pharmacology
 Please wait while we load your content...
Please wait while we load your content...